6.3 Create the alignment
In this section we’ll download relevant files and use the TCoffee software to create the alignment. We’ll add structure files to the PSI-Coffee method in order to provide information for structural alignment and annotations.
- Sequence files: we’ll use the same sequences that appear in Walls et al. (2020)
- Structure files: Protein Data Bank files 3D coordinates
6.3.1 Step 1: download sequence files
TASK
Download the sequence files listed in table @ref(tab:“Spike Glycoprotein S accession codes”) from section 6.2 above.
Files should be saved in the simple “fasta” format within a single text file.
Option 1: Retrieve previously saved sequences in a ready-to-use file: sarbecos.fasta
Option 2: Use NCBI “Batch Entrez” with the Protein database.
Create a list with accession files, for example using the following (Copy/Paste) code taken from the table above to create a text file named sarbecos.list.
echo "YP_009724390.1
QHR63300.2
AAP13441.1
AAP13567.1
AAS00003.1
AAV97988.1
AAV91631.1
ALK02457.1
AGZ48828.1
AVP78042.1
AVP78031.1
Q3I5J5.1
ACU31032.1" > sarbecos.listAlternatively download a premade list file: sarbecos.list
Then use that list on the Batch Entrez web site (see right hand side red arrow on figure 6.2.)
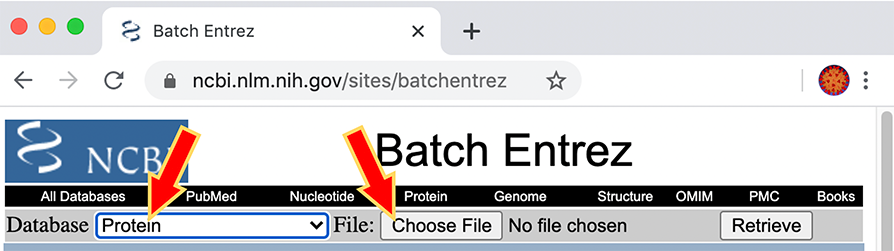
Figure 6.2: Details: Batch Entrez. Select Protein database and upload file list of accession codes.
Batch Entrez Instructions from the “Batch Entrez” web site:
“Given a file of Entrez accession numbers or other identifiers, Batch Entrez downloads the corresponding records.”
- Start with a local file containing a list of accession numbers or identifiers
- Select the database corresponding to the type of accession numbers or identifiers in your input file
- Use the Browse or Choose File… button to select the input file
- Press the Retrieve button to see a list of document summaries
- Select a format in which to display the data for viewing, and/or saving (Choose fasta)
- Select ‘Send to file’ to save the file.
6.3.2 Step 2: download structure files
We’ll download two Protein Data Bank (PDB) files in PDB format for the complete spike protein with code 6VXX and 6VYB. Since some portions of these structures are missing (too flexible to be dectected) we’ll add 2 other structures for the “receptor-binding domain complexed with its receptor ACE2” for SARS-CoV-2: 6LZG, and for SARS-CoV: 2AJF.
Note: for simplicity PDB files could be omitted.
Files can be downloaded from the web site or from the following command lines with either wget (“Web get” - not installed on MacOS by default) or curl (“Copy URL” - requires redirect):
# Commands with curl - (Copy URL)
curl http://files.rcsb.org/download/6VXX.pdb > 6vxx.pdb
curl http://files.rcsb.org/download/6VYB.pdb > 6vyb.pdb
curl https://files.rcsb.org/download/6LZG.pdb > 6lzg.pdb
curl https://files.rcsb.org/download/2AJF.pdb > 2ajf.pdb
# Alternate wget commands - (Web get)
wget http://files.rcsb.org/download/6VXX.pdb
wget http://files.rcsb.org/download/6VYB.pdb
wget https://files.rcsb.org/download/6LZG.pdb
wget https://files.rcsb.org/download/2AJF.pdbPlease not the exact writing (upper/lower case) of the files as they are saved.
Files should be saved in the same directory as the sequence file save previously.
6.3.3 Step 3: create alignment
Here we’ll use docker with a Docker image issued by the TCoffee authors at https://hub.docker.com/r/cbcrg/tcoffee from the Comparative Bioinformatics Centro de Regulacio Genomica (Centre for Genomic Regulation) hence CBCRG group. There is no information on the Docker Hub about the image itself but it is fully functional and its automated maintenance is detailed on the Release Building Procedure page.
TCoffee needs to be connected to the Internet to access the BLAST server on EBI and PSI databases. Therefore the docker command needs to provide a bridge to the Internet connection of the local computer. This is accomplished with the --net=host option. Other options mean: -it= interactive and terminal; --rm= delete the container when the job is done; -v= provide access to the current directory and map it to /data within the container; -w= define the default working directory within the container.
TASK
Make sure that the terminal is set to the directory containing both the sequence and structure files. Use pwd and ls to verify that this is the case.
We’ll now “plunge into” the docker container.
The prompt will change from $ to #.
Note: On HTML version of this document (but not PDF) we’ll be reminded that we are within the container by the blueish background behind the lines of command code.
The files used are named:
sarbecos.fasta: multiple sequence file in fasta format in the desired order for final output.*.pdb: 3D coordinate files in PDB format.
TCoffee command to compute the alignment used:
Command Details:
t_coffee: activate the TCoffee softwaresarbecos.fasta: multiple sequence file-outorder=input: keep the order of sequences as listed within the fasta file-seqnos: provide numbering on final output-pdb 6vxxA 6vybA 2ajfE 6lzgB: last letter added to PDB codes represents chain ID-mode psicoffee: choose running mode
6.3.4 Results
The complete alignment is shown in Appendix A.
Compute time:
On a Macbook Pro with 4 cores the computation will take about 20 minutes. Most of this time is dedicated to waiting for the live database connections as the reported local CPU time was only 1.22 second.
When using TCoffee via a web server15 it may take 48hrs or more to obtain a result.
References
Walls, A. C., Y. J. Park, M. A. Tortorici, A. Wall, A. T. McGuire, and D. Veesler. 2020. “Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein.” Cell 181 (2): 281–92. https://doi.org/https://dx.doi.org/10.1016/j.cell.2020.02.058.